

Protein Characterization using MMS
Proteins play a crucial role in most biological processes, and biochemical characterization of proteins enables researchers to investigate the relationship between structure and function. Spectroscopic techniques like Circular Dichroism (CD) and Fourier Transform Infrared Spectroscopy (FTIR) are used to measure protein secondary structure, which is essential for protein characterization. Primary structure measurement and protein sequence analysis, normally performed using mass spectroscopy, are useful for protein identification. CD and FTIR are used to analyze protein structure biology and protein stability, e.g. generating safer and more stable biologic drugs through biologics characterization. However, for reasons such as buffer interference and lack of sensitivity, there are limitations to the ability of the technologies to provide adequate functional characterization of biologic drugs.
These traditional technologies were, until now, among the only ones used for protein analysis and peptide analysis, which include secondary structure determination. The formation of these structural elements are fundamental events in protein folding structure and subtle but critical changes that can signal a protein misfolding issue. These small changes can affect product development and viability and are often not detected by CD and FTIR. Thus, these traditional techniques are not optimal for biophysical characterization of proteins in the development of pharmaceuticals.
An advanced IR-based technology called Microfluidic Modulation Spectroscopy (MMS) has recently emerged as a promising physicochemical characterization tool for biomolecule characterization. In particular, within a biotherapeutic development workflow, MMS can quantify and characterize the higher-order structure of proteins and other biomolecules across a range of measurements including stability, aggregation, and similarity without interference from excipients and at formulation concentration from 0.1 to >200 mg/mL. This technology, developed by RedShiftBio, provides valuable insights into the molecular mechanisms of proteins and can be relied on for lead optimization as well as quality assurance of biopharmaceuticals.

Learn how you can save time and money in your biotherapeutic development process.
Access relevant webinars and app notes now!

Why is Protein Characterization Important?
Microfluidic Modulation Spectroscopy (MMS) is a novel approach to spectroscopy that combines the advantages of microfluidic technology and infrared spectroscopic techniques to offer a superior protein characterization method. Central to MMS is the microfluidic flow cell that is used to control the flow of protein samples, modulating between sample and buffer for real-time background subtraction. The technology also utilizes a tunable Quantum Cascade Laser that is at least 1,000 times more intense than conventional light sources, allowing for a maximal signal-to-noise ratio resulting in ultra-high sensitivity.
MMS is the technology behind Aurora TX, a next-gen, fully-automated protein analyzer. MMS scientists have developed a vast protein library referenced within Aurora’s integrated delta software, to increase the relevance and accuracy of data processing by enabling the use of appropriate model proteins to analyze samples with unknown structures.
What is Microfluidic Modulation Spectroscopy?
Microfluidic modulation spectroscopy has a wide range of applications in the field of protein characterization. One of the most notable applications of this technology involves the upstream development and downstream quality assurance of biologic drugs, or biopharmaceuticals. MMS can perform protein characterization assays applied to drug target discovery such as protein discovery and antibody discovery, biological drug development (biologics development), and the formulation of protein therapeutics. In this field, MMS can help researchers select candidates that are least likely to misfold and most likely to be stable and survive the rigorous drug validation pathway. Some specific examples of biomolecule analysis in which MMS can provide valuable insights and save time and money in development include:
- Monoclonal antibody (mAbs) characterization
- Peptide analysis / peptide characterization
- Antibody drug conjugate characterization or ADC characterization
- Adeno-associated virus characterization or AAV characterization
- Biosimilar characterization
- Vaccine characterization
- Characterization of recombinant proteins
- Membrane protein analysis
- Ligand binding protein analysis
Applications of Microfluidic Modulation Spectroscopy
Microfluidic Modulation Spectroscopy is a promising method for the characterization of proteins and is considered one of the most sought-after protein characterization techniques available. MMS helps researchers build better biopharmaceuticals, faster and more cost-effectively. Its ability to provide rapid and automated measurements of protein structure, stability, and concentration without buffer interference, and to ultra-sensitively quantify previously undetectable structural changes in biomolecules is unmatched in the industry. This technology can interrogate a wide range of molecules, with an ever-growing protein database, or library of reference proteins. As the technology continues to evolve, MMS will likely become an increasingly important part of a researcher’s biophysical toolkit.
To learn more about MMS, download our resources or contact us today!
Frequently Asked Questions
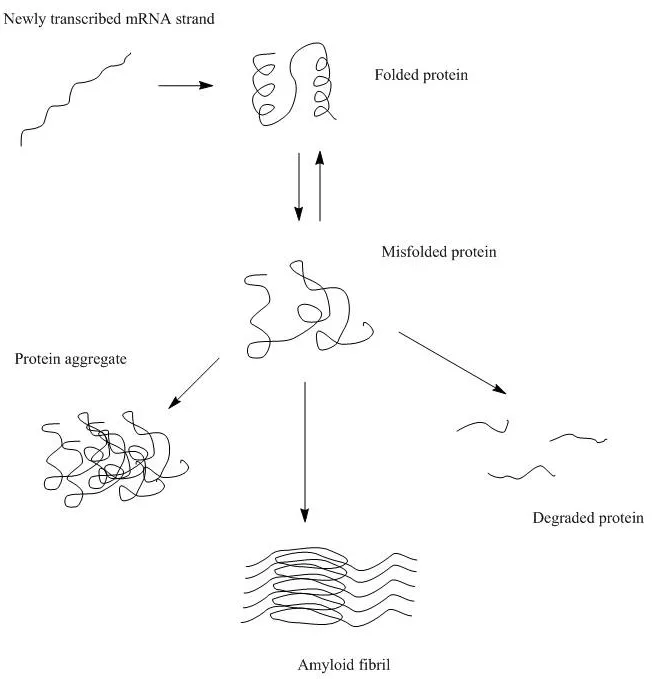
What causes protein aggregation?
The direct cause of protein aggregation results from interactions between individual proteins that become thermodynamically favored. There are two main sources of such driving force behind protein aggregation:
- The surface of proteins loses interaction with solvents (i.e., water molecules) and consequently exposes the hydrophobic groups that can interact with the surface of another protein undergoing the same process. Aggregates formed by such surface interactions are called colloidal aggregates.
- Mis-folded proteins fail to maintain the native structure and expose the inner hydrophobic pockets. The exposed hydrophobic pockets, primarily composed of β sheets, are prone to re-form beta interactions indiscriminately with other intra- and inter-molecular β sheets. Aggregates formed by these intermolecular β-sheets are called structural aggregates.
Why is protein structure important?
Protein structure in general is important for the protein function/ activity on the one hand and stability/ aggregation tendency on the other hand. Both the global protein structure, and 3D arrangement as well as local structural motifs and interactions determine those protein properties.
Resources:
Protein Analysis Tools: Higher Order Structure of Insulin | RedShiftBio
Using MMS to Measure Buffer-Induced Structural Changes in an Alpha-helix Rich Enzyme| RedShiftBio
Measuring Buffer-Induced Structural Changes in a Beta-sheet Rich Protein using MMS |RedShiftBio
Which protein structure is most stable?
Protein stability is dictated by the type of interactions that help the folding of the entire protein molecule. The primary structure of proteins is the most stable since the protein sequence is held together by covalent bonds between individual amino acids. Some of the tertiary structures that are held together by disulfide bonds are the most stable other than the primary sequence of the protein. Among the non-covalently bound structures, protein secondary structure is the most stable due to the predominant hydrogen bonding interactions.
What causes proteins to lose stability?
Unavoidable changes in temperature, pH or just moving the sample can trigger, for example, oligomerization or unfolding processes which can decrease the stability of the protein towards further stresses. The mechanisms that lead to protein instability can be diverse. The different mechanisms that can lead to a loss of stability in monoclonal antibodies are summarized in a review by Le Basle et al. (Le Basle, Yoann, et al. Physicochemical stability of monoclonal antibodies: a review." <em>Journal of Pharmaceutical Sciences</em> 109.1 (2020): 169-190.)
Is all structural change in proteins reversible/irreversible?
Structural changes in protein can be either reversible or irreversible. Changes in protein secondary structure are more likely to be irreversible as it is usually associated with loss of hydrogen bonding interactions. One example is beta-amyloid formation, which causes disease-related irreversible protein aggregation. However, lyophilization has been shown to cause reversible changes in protein secondary structure by increasing the beta-sheet content and decreasing the alpha-helix content (Klibanov et al. Lyophilization-induced reversible changes in the secondary structure of proteins. Proc Natl Acad Sci.1995;92(24):10969-76.) In addition, changes in tertiary or quaternary structures are more likely to be reversible. For example, aggregations due to the surface interaction or self-association of proteins that potentially alter the globular structure of the proteins and hence affect their activity or function, can be fixed by changing the formulation to tamper the surface interactions and separate the protein monomers.
